Definition and epidemiology
Dent’s disease (OMIM #300009) refers to a heterogeneous group of X-linked disorders that have previously been reported as X-linked recessive nephrolithiasis, X-linked hypercalciuric hypophosphataemic rickets, or idiopathic low-molecular-weight proteinuria with hypercalciuria and nephrocalcinosis . The disease is characterized by manifestations of proximal tubule (PT) dysfunction associated with hypercalciuria, nephrolithiasis, nephrocalcinosis, and progressive renal failure . Low-molecular-weight (LMW) proteinuria represents the most consistent manifestation of Dent’s disease, detected in almost all affected males and obligate female carriers.
Clinical description
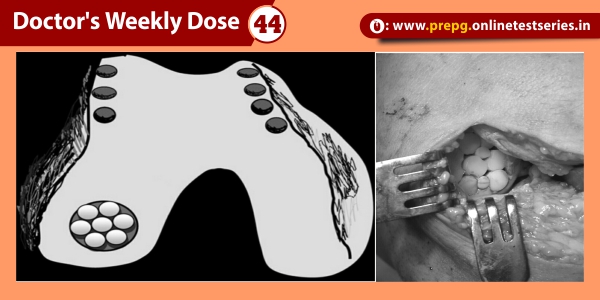
Dent’s disease is characterized by PT dysfunction and LMW proteinuria, associated with hypercalciuria, nephrolithiasis, nephrocalcinosis, and progressive renal failure. Dent’s disease may also be associated with aminoaciduria, phosphaturia, glycosuria, uricosuria, kaliuresis, and impaired urinary acidification, and is often complicated by rickets or osteomalacia .These features are generally found in males only, who may have manifestations of the disease from early childhood. These patients may present with bone pain and difficulty in walking due to rickets, or symptoms of renal stones such as abdominal pain and haematuria. Occasionally, patients are referred as a result of the fortuitous discovery of biological manifestations of PT dysfunction, including LMW proteinuria. LMW proteinuria, which is characterised by the excretion of proteins such as α1 and β2 microglobulins, retinol-binding protein (RBP), Clara cell protein, and vitamin D binding protein, is found in approximately 99% of affected males.
Some patients with Dent’s disease have been observed to have extra-renal manifestations such as mild intellectual impairment . hypotonia and cataract, and such patients have been reported to share mutations in OCRL1 with the oculo-cerebrorenal syndrome of Lowe . The occurrence of these extra-renal manifestations with mutations relating to Lowe syndrome is referred to as Dent disease 2 . To date, around 20 patients with Dent disease 2 have been reported, all of whom have hypercalciuria and LMW proteinuria. In addition, these patients may also have nephrocalcinosis, nephrolithiasis, haematuria, hypophosphataemia and/or renal insufficiency. Only a minority (approximately one-fourth) of these patients have been observed to have mild intellectual deficit, hypotonia and sub-clinical cataract.
Genetics
Dent’s disease may be caused by either inactivating mutations in CLCN5(OMIM #300008), which is located on chromosome Xp11.22 and encodes a 746 amino-acid electrogenic Cl–/H+ exchanger (ClC-5) , or theOCRL1 gene, which is located on chromosome Xq25 and encodes the phosphatidylinositol 4,5-biphosphate 5 -phosphatase OCRL1. ClC-5 contains 18 α-helices, with two phosphorylation and one N-glycosylation sites. Structural studies have revealed that the protein forms diamond-shaped homodimers composed of two repeated halves that span the membrane in opposite orientations. Each subunit has its own pore responsible for the selective coupling of the Cl– flux to H+ counter-transport .
Approximately 40% of patients with Dent’s disease do not have CLCN5mutations, even though they are clinically indistinguishable from those that have CLCN5 mutations. Twenty of these patients have been reported to have OCRL1 mutations although it is important to note that none of these had the severe cataracts or intellectual deficit that is typically found in patients with Lowe syndrome. Consistent with these phenotypic differences, it is interesting to note that the OCRL1mutations associated with Dent disease 2 do not overlap with those causing Lowe syndrome.
Pathophysiology
The complex phenotype of Dent disease 1 is probably explained by the predominant expression of ClC-5 in the PT segments, with more discrete expression in the thick ascending limb (TAL) of Henle’s loop and the α-type intercalated cells (IC) of the collecting ducts of the kidney . In PT cells, ClC-5 co-distributes with the vacuolar H+-ATPase (V-ATPase) in early endosomes , which are responsible for the reabsorption and processing of albumin and LMW proteins that are filtered by the glomerulus (Figure ). These vesicles belong to the receptor-mediated endocytic pathway, which involves the multiligand receptors, megalin and cubilin, located at the apical brush border of PT cells. Progression along the endocytic apparatus depends on endosomal acidification, driven by the V-ATPase and requiring a parallel Cl–conductance to maintain electroneutrality. It has long been assumed that ClC-5 could provide such an electrical shunt to neutralize the H+gradient. Accordingly, the loss of the endosomal Cl– conductance mediated by ClC-5 would impair vesicular acidification, causing dysfunction of PT cells. Two independent strains of ClC-5 knock-out (KO) mice have been generated, which both recapitulate the major features of Dent’s disease including LMW proteinuria and other manifestations of PT dysfunction. Furthermore, in vitro experiments have shown a decreased acidification of early endosomes in ClC-5-deficient mice .
Treatment
In the absence of therapy targeting the molecular defect, the current care of patients with Dent’s disease is supportive, focusing on the prevention of nephrolithiasis. Thiazide diuretics can be used to treat hypercalciuria . although significant adverse events, including hypovolemia and hypokalemia related to the primary tubulopathy, have been reported .Similarly, treatment of rickets with vitamin D must be cautious since it may increase hypercalciuria. Studies performed on ClC-5-deficient mice suggest that long-term control of hypercalciuria by a high citrate diet delays progression of renal disease even in the apparent absence of stone formation .